

植物各个部位(如根、茎、叶、花和果实)栖居着大量高度多样的微生物(即植物微生物组),包括细菌、真菌、古菌和原生生物等,这些微生物与宿主植物共同形成一个“共生总体”,彼此相互作用、共同进化。植物微生物组在植物的生长发育、养分吸收、生物及非生物胁迫抗性等方面发挥重要作用,但目前对于作物微生物组群落构建的基本生态过程及植物-微生物互作的微生态机制还缺乏系统的认识。
本研究在河南许昌(潮土)和云南曲靖(红壤)旱地农田设置了包括7个不同施肥处理的田间试验,研究了玉米-小麦/大麦轮作下土壤(根际及非根际土壤)及多个植物部位所代表的生态位(根表、根内、叶表、叶内)共684个样品中细菌的群落构建机制。
结果表明,在土壤-植物连续体上,微生物组群落构建主要由宿主选择(即生态位和作物物种差异)决定,而地点或施肥措施的影响较小。从土壤到植物表面再到植物内部,宿主选择效应逐渐增加而细菌群落的多样性及网络复杂度相应降低,在叶内具有较强的宿主选择效应。溯源分析进一步揭示了作物微生物组主要来自土壤环境并逐步被不同的植物部位富集和过滤。此外,我们发现作物微生物组由少量的优势类群(约占0.5%的细菌ZOTUs)主导,芽孢杆菌纲(Bacilli)和甲基杆菌科(Methylobacteriaceae)分别被鉴定为麦类(大麦和小麦)和玉米的重要指示生物类群。
这些研究结果为作物微生物组群落构建的宿主选择、潜在来源及富集过程提供了综合性的实验证据,并为未来基于土壤、作物微生物组精准调控的可持续农业管理提供了重要信息。
大量高度多样的微生物生活在植物的各个部位,包括根、茎、叶、花和果实。植物及其微生物组共同形成一个“共生总体”(holobiont),彼此相互作用、共同进化。与人体微生物组类似,植物微生物组作为植物的第二个基因组,与宿主植物生长和健康紧密相关。充分利用和发挥植物微生物组功能以提高作物产量是未来农业可持续发展的潜在途径,但其前提条件是提高我们对驱动微生物组群落构建的生态过程的基本认识。
宿主植物为微生物群落提供了多种微生境,包括多种植物器官,植物外部组织(外生菌),及内部组织(内生菌)。土壤是宿主微生物来源的一个极其丰富的微生物资源库。已有的研究表明,植物不同部位的微生物群落明显不同,微生物在植物不同部位的成功定殖,需要其有能力克服不同微生境中的宿主免疫及非生物压力。对植物根际及根系微生物组的研究发现,微生物组群落构建受多种因素决定,包括植物遗传多样性、生长时期、土壤类型和土壤养分状况等。但目前对土壤-植物连续体上,包括非根际、根际和植物表面及内部各部位中微生物组群落构建机制的理解仍然有限,尤其对于田间不同施肥管理条件下生长的作物。
除了来自宿主的生物压力,农业生态系统中的微生物群落经常还受到不同气候、土壤性质、及农业管理措施的影响。例如,大量的研究发现土壤性质(如pH和养分)和农业管理制度(如施肥措施及耕作强度)可以显著影响土壤及根系微生物组的群落组成。这些研究表明作物相关的微生物组是由多种生物及环境因素共同决定的,然而目前的大部分研究只关注了其中的一个或者少数几个因素。对于植物不同部位、宿主植物种类、土壤因素及农业管理扰动等多种因素如何共同影响作物微生物组群落构建,我们仍然缺乏系统的认识。大量研究表明,通过秸秆还田、硝化抑制剂、生物炭和微生物菌剂应用等可以有效提高土壤肥力及植物生产力。因此,本研究在河南许昌(潮土)和云南曲靖(红壤)旱地农田设置了包括7个不同施肥措施的田间试验,研究了玉米-小麦/大麦轮作下土壤(根际及非根际土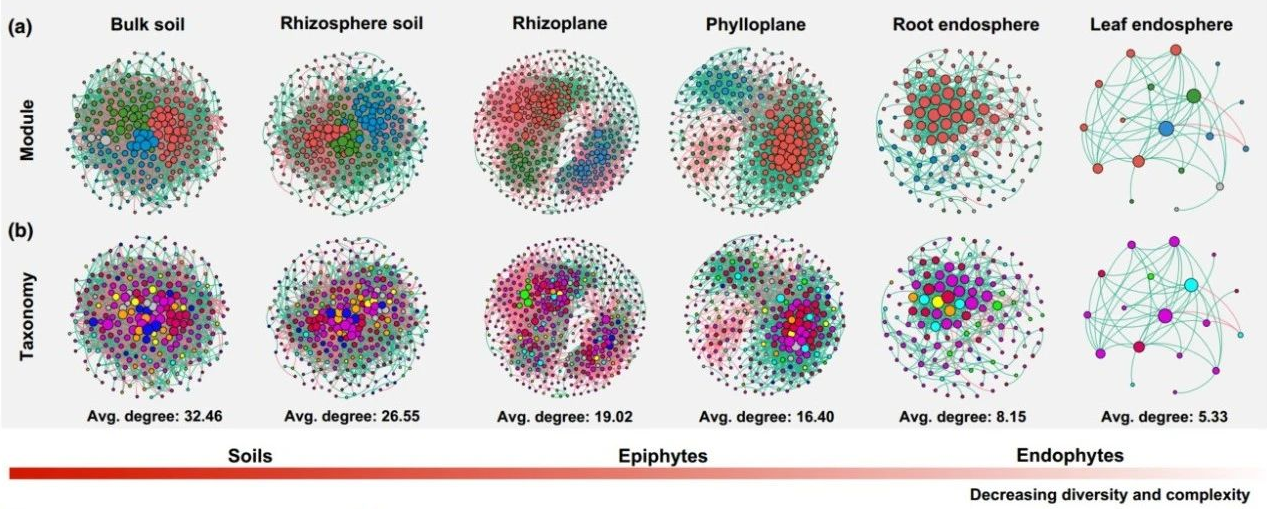 壤)及多个植物部位所代表的生态位(根表、根内、叶表、叶内)共684个样品中细菌的群落构建机制。本研究的目的包括:(1)评估宿主(不同部位生态位及作物种类)及环境(地点及施肥措施)因素在根际、叶际、及内生环境中如何共同影响作物微生物组群落构建及网络共现模式;(2)探究土壤-植物连续体上作物微生物组的潜在来源及关键微生物类群。我们的科学假设是:(1)宿主效应和环境效应对作物微生物组群落构建的相对贡献从土壤到植物表面到植物内部会逐渐转变,由于逐渐增加的宿主效应,微生物群落多样性及网络复杂度会相应的逐渐降低;(2)从非根际土壤到植物内部,作物微生物组会被逐步富集和过滤,植物内生环境可能会从邻近的物种库选择主要的微生物类群。
壤)及多个植物部位所代表的生态位(根表、根内、叶表、叶内)共684个样品中细菌的群落构建机制。本研究的目的包括:(1)评估宿主(不同部位生态位及作物种类)及环境(地点及施肥措施)因素在根际、叶际、及内生环境中如何共同影响作物微生物组群落构建及网络共现模式;(2)探究土壤-植物连续体上作物微生物组的潜在来源及关键微生物类群。我们的科学假设是:(1)宿主效应和环境效应对作物微生物组群落构建的相对贡献从土壤到植物表面到植物内部会逐渐转变,由于逐渐增加的宿主效应,微生物群落多样性及网络复杂度会相应的逐渐降低;(2)从非根际土壤到植物内部,作物微生物组会被逐步富集和过滤,植物内生环境可能会从邻近的物种库选择主要的微生物类群。
基于所有样本的NMDS排序及PERMANOVA分析结果表明,细菌群落变异主要由部位生态位 (R2 = 39.8%, P < 0.001) 及宿主种类 (R2 = 7.8%, P < 0.001) 解释,然后是地点 (R2 = 2.7%, P < 0.001) 和施肥措施 (R2 = 0.4%, P = 0.07) (Fig. 1a)。不同施肥措施对第一个作物季(2016年玉米季)(R2 = 0.7%, P = 0.83) 的细菌群落结构没有显著影响,但是对第二季(2017年小麦/大麦季)(R2 = 0.6%, P = 0.03) 及第三季(2017年玉米季)(R2 = 1.2%, P < 0. 001)具有显著影响。

宿主在塑造作物微生物组方面占主导地位;
a 基于所有样品(n = 684)、2016年玉米样品(n = 216)和2017年玉米样品(n = 252)以及2017年小麦/大麦样品(n = 216)的加权UniFrac距离矩阵的非度量多维尺度排序(NMDS)分析。XC,许昌站点;QJ,曲靖站站;
b 基于2017年样本中每个菌群生态位的加权UniFrac距离的NMDS排序分析(n = 468)。HEEI,宿主-环境效应指数(HEEI 值越高表示宿主物种效应越大)。Env. effect,环境效果。BS,散土体土壤;LE,叶内圈;P,叶面;RS,根际土壤;R,根面;RE,根内圈。
我们进一步定义HEEI指数(Host-environment effects index,宿主-环境效应指数,较高的HEEI代表较高的宿主效应)来评估宿主及地点对不同部位细菌群落的相对贡献。我们发现从土壤(非根际土壤: 0.13, 根际: 0.34)到植物表面(根表: 3.36, 叶表: 0.83)到植物内部(根内: 4.93, 叶内: 10.79),HEEI值逐渐上升,表明从土壤到植物表面到植物内部宿主效应逐渐升高,在叶内具有最强的宿主效应 (宿主种类解释了60.4% 的群落变异) (图1b)。相应地,从土壤到植物表面到植物内部,环境效应逐渐降低。地点分别解释了根际及非根际土壤微生物群落48.3% 及 56.5%的变异(图1b,表 1)。此外,施肥措施对根际 (R2 = 6.7%, P < 0. 001), 非根际土壤 (R2 = 4.6%, P = 0.041), 叶内 (R2 = 4.4%, P = 0.011), 及根表 (R2 = 2.7%, P = 0.049)的细菌群落具有显著影响,但是与作物种类及地点相比这种影响非常小。

a HEEI,宿主-环境效应指数(HEEI = 作物物种的相对贡献/场地的相对贡献,基于嵌套 PERMANOVA 分析)。HEEI 值越高表示寄主物种效应越大。
为了进一步表征宿主选择效应对作物微生物组的影响,我们评估了土壤-植物连续体上细菌群落的alpha多样性及网络共现模式。结果表明,细菌丰富度指数(Chao1)及网络复杂度从土壤(平均连接度,非根际土壤: 32.46, 根际: 26.55)到植物表面(根表: 19.02, 叶表: 16.40)到植物内部(根内: 8.15, 叶内: 5.33)逐渐降低,并以叶内最低(图2a-c)。微生物网络的“中心节点” (节点具有较高的连接度 (> 60) 及紧密中心性 (> 0.3))数量从土壤到植物表面到内生逐渐降低(图2d,表2)。土壤及植物不同部位的微生物网络物种组成具有明显差异,土壤微生物网络中有更多的酸杆菌门类群,而在植物部位中有更多的厚壁菌门类群(图2b)。值得注意的是,厚壁菌门肉杆菌属的微生物类群是根表细菌网络的中心节点(图2b)。此外,我们在叶表和根表的微生物网络中发现了较高的模块化程度及平均路径距离(图2a)。

作物宿主对减少细菌多样性和网络复杂性有很强的影响;
a,b 基于 2017 年收集的样本(n = 468)沿土壤-植物连续体的细菌共现网络;
c 基于所有样本的不同生态位中的细菌 alpha 多样性(n = 684)。endo,内圈;
d 不同生态位中细菌网络“枢纽节点”的分布模式。

a 节点倾向于分化为不同网络模块的程度;
b 节点倾向于聚集在一起的程度;
c 网络路径距离是网络内两个节点之间最短路径的长度;
d 关键核心节点被定义为在网络中具有高的度数(> 60)和紧密中心性(> 0.3)的节点。
植物微生物组来源模型(Source Model of Plant Microbiome, SMPM)表明,作物相关细菌群落主要来源于非根际土壤并被不同的植物部位逐步过滤。根表、根内及叶内生境可能从邻近的物种库选择主要的微生物类群,已知的潜在来源> 78%,根内生菌是叶内生菌的主要潜在来源(图3a)。对每种作物分别进行的溯源分析也发现了类似的特征。

作物相关细菌群落主要来自土体土壤,并通过不同的部位生态位逐渐富集和过滤;
a 植物微生物组 (SMPM) 的溯源模型显示了基于 2017 年收集的样本(n = 468)的作物相关细菌群落的潜在来源。U,未知源(线的粗细相当于源的贡献);
b 火山图显示了与土体土壤相比,每个部位生态位中作物相关细菌微生物群的富集和减少模式。每个点代表一个零半径操作分类单元(ZOTU;RA,相对丰度 > 0.1%,总共 6744)。每个红点代表一个单独的富集 ZOTU,一个绿点代表一个单独的减少ZOTU。沿 y 轴的位置表示与土体土壤相比丰度的倍数变化,x 轴代表平均 ZOTU 丰度(每百万计数,CPM)。DI,耗尽指数;DSI,相异指数;All,每个生态位中总 ZOTU 的数量(RA > 0.1%)。ns,不显著;
c 维恩图显示了不同生态位中显著富集的 ZOTU 和减少的 ZOTU 内中共享和特有的细菌ZOTU。对于这些共有的差异 ZOTU,仅显示了前三个的物种分类。
差异分析表明以肠杆菌科 (40 ZOTUs), 假单胞菌科 (36 ZOTUs) 及甲基杆菌科 (30 ZOTUs) 为代表的310个ZOTUs (占总 6744 ZOTUs的4.6%)在4个植物部位(根表、根内、叶表和叶内)被显著富集 (图3b,c)。而以噬几丁质菌科(101 ZOTUs)为代表的1427个ZOTUs(占总ZOTUs数的21.2%)在这4个植物部位显著降低(图3b,c)。其次,根表及叶表富集的特异性ZOTU最多(图3b,c)。我们进一步通过定义DI(Depleted index, 下调指数)及DSI(Dissimilarity index, 不相似度指数)来评估微生物群落从非根际土壤到不同植物部位的物种过滤及选择过程。发现DI指数从根际 (0.08) 到植物表面 (0.90-1.32) 到内部 (1.60-2.12)逐渐升高, 在叶内发现最高的 DI 及 DSI 指数值 (图3b)。
为了更好的表征作物物种水平上的宿主效应,我们分别鉴定了每种作物(只包括叶片及根系样品)的优势细菌类群(dominant taxa, 在至少60%的植物样品中出现且相对丰度≥ 0.1%)及指示类群(基于LEfSe)。虽然在每种作物中都发现了超过10000个ZOTUs,但是只有43 (0.2%), 91 (0.7%), 及 65 (0.6%)个 ZOTUs分别被鉴定为玉米、小麦、及大麦的优势细菌类群,这些ZOTUs占每种作物总序列数的~50%(31.1, 62.5, 及 49.1%)(图4a-c)。对于所有3种作物,这些优势的ZOTUs主要属于γ变形菌纲,在每种作物的优势类群中相对丰度占39%-50%(图4a-c),并以肠杆菌科为主(图4a-c)。芽孢杆菌纲是小麦(19.0%)及大麦(9.0%)的优势类群,但在玉米中只占0.8%(图4a-c)。LEfSe分析进一步表明芽孢杆菌属、乳杆菌科(属于芽孢杆菌纲)、及甲基杆菌科分别是大麦、小麦、及玉米的指示生物类群。随机森林模型分析和优势类群的功能预测分析表明,γ变形菌纲及芽孢杆菌纲分别是玉米和大麦/小麦产量的主要预测因子,且玉米样品(3.8%)比小麦(0.8%)及大麦(0%)样品具有更多的甲基营养功能组,进一步表明甲基杆菌科是玉米的重要指示类群。


作物优势类群的系统发育树、物种分类组成和分布模式;
a 玉米的优势分类群的鉴定(仅考虑叶和根区,n = 312);
b 小麦优势类群的鉴定(n = 72);
c 大麦优势类群的鉴定(n = 72);
优势类群被定义为平均相对丰度≥ 0.1% 的零半径操作分类单位 (ZOTU),并且存在于> 60%的所有样本中。三种作物中占优势类群总序列数 < 2% 的低丰度类群被归为“其他”。
本研究系统揭示了宿主和环境因素对玉米、小麦和大麦微生物组群落构建的相对贡献,证实了在土壤-植物连续体上,微生物组群落构建主要由部位生态位及宿主种类决定,而地点(及地点相关的土壤和气候因子)或施肥措施的影响较小。本研究进一步揭示了从土壤到植物表面再到内生,宿主选择效应逐渐增加并能够显著降低细菌群落的多样性及网络复杂度。这些发现促进了我们对不同环境选择压力下玉米-小麦/大麦系统中细菌群落构建的认识,并突出了宿主选择效应的重要性。
其次,本研究为植物微生物组的逐步过滤及富集过程提供了实验性证据,证实了微生物群落从非根际土壤经根际、根表逐步被富集到作物不同部位。相比于土壤,植物组织显著富集了肠杆菌科(Enterobacteriaceae),假单胞菌科(Pseudomonadaceae)和甲基杆菌科(Methylobacteriaceae)的细菌。作物内生环境可以从邻近的物种库选择主要的微生物类群,根内生菌是叶内生菌的主要潜在来源。研究还进一步揭示了作物微生物组由少量属于γ变形菌纲的优势类群(约占0.5%的细菌ZOTUs)主导,芽孢杆菌纲(Bacilli)和甲基杆菌科(Methylobacteriaceae)分别是麦类(大麦和小麦)和玉米的重要指示生物类群。这些研究结果丰富了植物微生物组群落构建的生态进化理论,为未来基于土壤、作物微生物组精准调控的可持续农业管理提供了重要的信息。
田间施肥处理于2016年春天开始进行,在两个研究地点都包括7个不同的处理:(1)不施氮肥(Control),(2)常规施肥(N),(3)常规氮肥处理减氮20%(80%N),(4)减氮20%加秸秆还田(80%NS),(5)减氮20%加硝化抑制剂(80%NI),(6)减氮20%加叶际喷施固氮菌Klebsiella variicola W12(80%NKle),(7)减氮20%加生物炭和秸秆还田(80%NSB)。

对于叶片及根系样品,每个小区随机选取约5株玉米及20-30株小麦/大麦,对每株植物中上部的1-2片健康叶进行采集并立刻放入冰袋中。通过抖根法采集同株植物的根际土壤及根系。同时采集距离根系20cm外的表层土作为非根际土壤,每个小区进行5点取样混合均匀后作为一个生物重复。
将10-15g叶片及3-5g根(表面附着的根际土壤已被小心去除)放入带有缓冲液(0.1 M 磷酸钾缓冲液,含0.1% 甘油, 0.15% 吐温 80, pH 7.0)的蓝盖瓶中,在40 kHz下超声1min,然后在速度为200 rpm的摇床上震荡4min对微生物细胞进行收集。这个过程重复2-3次,然后将带有微生物细胞的缓冲液通过0.22 μm滤膜。使用PowerSoil DNA 提取试剂盒按照说明书对得到的滤膜进行DNA提取。
对于植物内生DNA,按上述方法处理后,将得到的叶片或者根系用无菌水洗净后进行表面消毒灭菌。步骤为先放入70%酒精中浸泡5min,再放入5.25%次氯酸钠溶液中浸泡5分钟,然后放入70%酒精中浸泡30秒,最后用无菌水清洗5次。使用液氮及无菌研钵对这些处理后的叶片及根系进行研磨,称取0.4g粉末样品使用PowerSoil DNA试剂盒进行内生DNA提取。采用799F/1115R引物对细菌16S rRNA V5-V6 区进行扩增。
Chao Xiong,Brajesh K. Singh, Ji-Zheng He, Yan-Lai Han, Pei-Pei Li, Li-Hua Wan, Guo-Zhong Meng, Si-Yi Liu, Jun-Tao Wang, Chuan-Fa Wu, An-Hui Ge, Li-Mei Zhang. Host selection shapes crop microbiome assembly and network complexity. New Phytologist. 2021, 229: 1091-1104. https://doi.org/10.1111/nph.16890