
近期,顶尖学术期刊《自然》在线连发三篇论文。这3篇论文里,由美国、意大利、丹麦科学家领导的多支团队从不同角度切入,找到了一个叫做AMBRA1的关键调控蛋白,并介绍了其调控周期蛋白D的机制,以及对癌症治疗的潜在意义。

https://www.nature.com/articles/s41586-021-03445-y

https://www.nature.com/articles/s41586-021-03474-7

https://www.nature.com/articles/s41586-021-03422-5
在细胞分裂过程中,细胞周期蛋白D结合并激活细胞周期蛋白依赖性激酶4(CDK4)和细胞周期蛋白依赖性激酶6(CDK6)。这些细胞周期蛋白-CDK4/6激酶给肿瘤抑制蛋白RB1、RBL1和RBL2添加磷酸基团,从而推动了细胞分裂。细胞周期蛋白D-CDK4/6激酶的不受控制激活是许多类型癌症发展的驱动力。
随着CDK4/6激酶的小分子抑制剂进入临床,人们对细胞周期蛋白D-CDK4/6生物学的兴趣越来越大。这些化合物的临床试验结果令人瞩目,证明它们有能力延长乳腺癌患者的生存时间。CDK4/6抑制剂帕博西尼(palbociclib)、瑞博西尼(ribociclib)和阿贝西利(abemaciclib)被批准用于治疗晚期乳腺癌。此外,这些药物正在几百项临床试验中对许多不同类型的癌症进行测试。
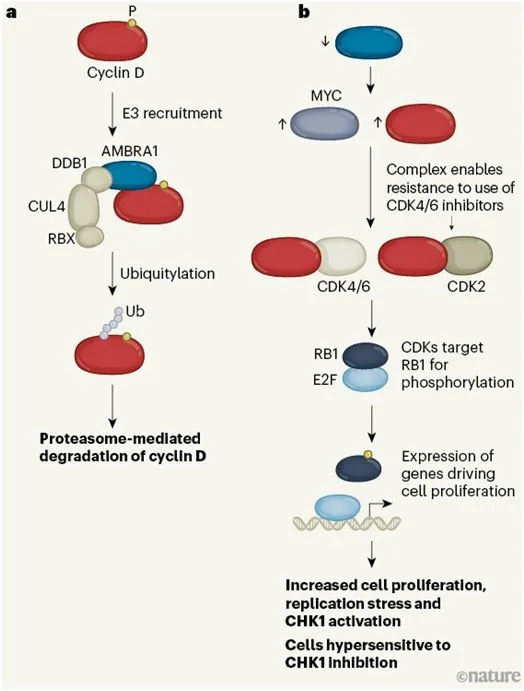
细胞周期蛋白D的破坏,图片来自Nature, 2021, doi:10.1038/d41586-021-00889-0。
自20世纪90年代发现细胞周期蛋白D以来,人们对其进行了深入研究,但它们在细胞周期中是如何降解的仍有争议。细胞周期蛋白D的羧基末端区域磷酸化触发这些蛋白被称为泛素-蛋白酶体系统的降解途径所破坏。在这个系统中,由泛素激活酶(E1)、泛素结合酶(E2)和泛素连接酶(E3)组成的级联活性,通过一个称为泛素化的过程,将几个称为泛素的小蛋白分子链附着在目标蛋白上。然后,这些泛素化的蛋白注定要在一种称为蛋白酶体的蛋白复合物中遭受降解。
最大的E3家族是cullin–RING连接酶(CRL)。CRL由一个cullin蛋白、一个RING蛋白(可招募E2)、一个衔接蛋白和许多不同的底物受体蛋白之一组成,其中底物受体蛋白负责将目标蛋白招募到E3复合物中。称为CRL1的E3的几种底物受体被认为与细胞周期蛋白D1的降解有关,而其他的E3底物受体则被认为靶向细胞周期蛋白D2和D3以便它们随后遭受蛋白酶体破坏。
此外,细胞周期蛋白D1被后期促进复合物(anaphase-promoting complex)泛素化,这种后期促进复合物是一种靶向几种细胞周期蛋白的E3复合物。与这些模型相反,其他研究则指出细胞周期蛋白D1的水平和稳定性不受这些蛋白耗竭的影响,这表明一些其他的E3调节着细胞周期蛋白D1的降解。
这三项新的研究指出细胞周期蛋白D1、D2和D3被称为CRL4的E3泛素化,随后遭受蛋白酶体降解,CRL4使用蛋白AMBRA1作为它的底物受体。人们已经知道AMBRA1在调节自噬方面具有关键作用,其中自噬是细胞降解受损细胞器或蛋白聚集物的过程。AMBRA1也已被确定为E3(包括CRL4)的底物受体。
通过一系列实验,利用包括细胞生物学、分子生物学和发育遗传学等领域的技术,这三项新的研究证实在正常细胞、癌细胞以及发育中的小鼠胚胎中,AMBRA1的耗竭会导致细胞周期蛋白D水平的上升。与具有正常数量AMBRA1的细胞相比,这导致了更大的RB1磷酸化和更多的细胞增殖。
第三项研究还显示,AMBRA1的耗尽会提高转录因子蛋白N-MYC的水平。这些研究人员之前已发现,AMBRA1调节一个相关的转录因子---c-MYC---的稳定性和活性。MYC家族蛋白可以上调细胞周期蛋白D和细胞周期蛋白E的表达,从而加速细胞周期的进展。
这些观察表明,AMBRA1可能作为一种肿瘤抑制蛋白发挥作用。事实上,只有AMBRA1编码基因的一个功能性拷贝的小鼠比具有这个基因的两个功能性拷贝的正常小鼠有更高的肺部、肝脏和肾脏肿瘤发生率。这三项新的研究提供了令人信服的证据来支持这一观点。
这些研究人员证实,AMBRA1基因在人类癌症中发生了突变。正如预期的那样,鉴于AMBRA1能够促进细胞周期蛋白D1的降解,他们报告说,人类肿瘤中AMBRA1的水平与细胞周期蛋白D1的水平呈反比关系。
肿瘤中AMBRA1的低水平与癌症患者的预后不佳有关。不论是AMBRA1在人类肿瘤细胞系中的实验性失活,还是在小鼠细胞中经过基因改造后发生促癌突变而导致的AMBRA1实验性失活,均会增加这些细胞在注射到小鼠体内后的肿瘤形成潜力。
此外,在Kras基因突变版本驱动的肺癌小鼠模型中,AMBRA1的基因剔除促进了肿瘤的形成,而且这些AMBRA1缺乏的肿瘤具有高于正常水平的细胞周期蛋白D。总之,这些研究表明,AMBRA1在正常情形下抑制细胞增殖,主要是通过阻止细胞周期蛋白D达到较高的水平。
第一项研究和第二项研还证实,AMBRA1的丢失以及细胞周期蛋白D的同时增加,导致人类肿瘤细胞对CDK4/6抑制剂的敏感性下降。有趣的是,这些作者报告说,在AMBRA1缺失的细胞中,细胞周期蛋白D1不是主要与CDK4/6合作,而是与细胞周期蛋白依赖性激酶CDK2形成一种具有催化活性的复合物,而且这种复合物对CDK4/6抑制剂不敏感。
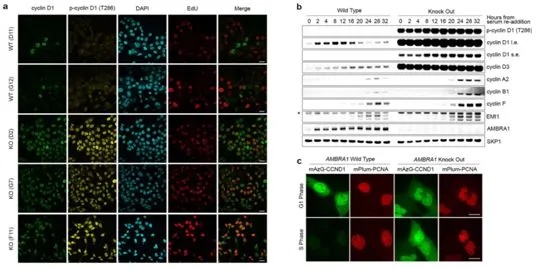
在正常细胞周期中,在营养剥夺和基因毒性胁迫后,AMBRA1的缺失可调节细胞周期蛋白D。图片来自Nature, 2021, doi:10.1038/s41586-021-03445-y。
第三项研究还表明,AMBRA1的丧失以及由此产生的细胞周期蛋白D(也可能还有其他蛋白,如c-MYC)的上升,引发了DNA损伤和复制压力(replication stress),这伴随着一种叫做CHK1的细胞周期检查点激酶(cell cycle checkpoint kinase, CHK)的激活。重要的是,这些作者报告说,AMBRA1缺失的癌细胞对CHK1抑制剂治疗不敏感,这表明存在靶向AMBRA1缺失肿瘤的潜在治疗机会。
这些令人兴奋的结果提出了几个关键问题。例如,AMBRA1水平的下降是否是人类肿瘤对CDK4/6抑制剂产生现有抵抗性和获得性抵抗性的基础?随着AMBRA1的耗尽,细胞周期蛋白D的上升是否是导致对CDK4/6抑制剂产生抵抗性的唯一因素?
对乳腺癌患者临床试验的分析显示,编码细胞周期蛋白D1的基因的额外拷贝的存在,或肿瘤中细胞周期蛋白D1信使RNA或蛋白的水平,与患者对CDK4/6抑制剂的反应之间没有关联。
事实上,第二项研究发现,经驱动后细胞周期蛋白D表达量高于正常水平的人类癌细胞并不完全再现AMBRA1耗尽时观察到的抑制剂抵抗性特征。也许其他受到AMBRA1调控的蛋白,如可以上调细胞周期蛋白E并激活细胞周期蛋白E-CDK2复合物的c-MYC,对治疗抵抗性有贡献。
第一项研究和第二项研究观察到在AMBRA1耗尽的细胞中形成抵抗CDK4/6抑制剂的细胞周期蛋白D-CDK2复合物,这很耐人寻味。这种“非典型”复合物以前被发现是对CDK4/6抑制产生获得性抵抗性的基础。这些作者推测,AMBRA1耗尽在某种程度上促进了这些细胞周期蛋白D-CDK2复合物的形成,这与细胞周期蛋白D水平的升高一起,导致了对CDK4/6抑制剂的抵抗性。第三项研究提出的一个特别令人兴奋的可能性是,CHK1抑制剂可用于治疗AMBRA1水平较低的对CDK4/6抑制剂有抵抗性的肿瘤。
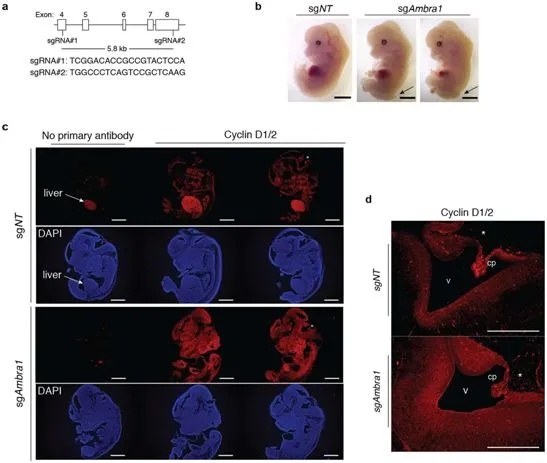
小鼠胚胎中的AMBRA1缺失导致细胞周期蛋白D水平升高。图片来自Nature, 2021, doi:10.1038/s41586-021-03474-7。
对AMBRA1在人类癌症中的作用开展进一步研究是有必要的。AMBRA1的肿瘤抑制功能主要是由细胞周期蛋白D1或c-MYC介导的,还是也由其他靶标介导的?不再产生RB1的肿瘤细胞不需要细胞周期蛋白D来促进细胞周期进展,因此,如果细胞周期蛋白D是AMBRA1的主要靶标,那么预计在产生RB1的肿瘤中会观察到AMBRA1的缺失。
此外,人类肿瘤中的AMBRA1缺失是否与影响细胞周期蛋白D羧基末端的突变相互排斥仍有待确定,这可能会使细胞周期蛋白D对AMBRA1介导的降解产生抵抗性。
另一个未解决的问题是,正如第二项研究报告的那样,为何AMBRA1的低表达与高水平的细胞周期蛋白D1相关,并与具有特定类型基因改变(如Kras突变)的肺部肿瘤的不良生存率相关。对于那些具有编码EGFR蛋白的基因突变版本或具有野生型Kras的肺部肿瘤则没有观察到这种影响。
不管这些问题的答案如何,这三项新的研究令人印象深刻,改善了科学家们对细胞周期进展机制的理解。
参考资料:
1.Daniele Simoneschi et al. CRL4AMBRA1 is a master regulator of D-type cyclins. Nature, 2021, doi:10.1038/s41586-021-03445-y.
2.Andrea C. Chaikovsky et al. The AMBRA1 E3 ligase adaptor regulates the stability of cyclin D. Nature, 2021, doi:10.1038/s41586-021-03474-7.
3.Emiliano Maiani et al. AMBRA1 regulates cyclin D to guard S-phase entry and genomic integrity. Nature, 2021, doi:10.1038/s41586-021-03422-5.
4.Charupong Saengboonmee et al. The path to destruction for D-type cyclin proteins. Nature, 2021, doi:10.1038/d41586-021-00889-0.